

Abstract
Introduction:
Diffuse large B-cell lymphoma (DLBCL) is an aggressive and heterogeneous disease that is characterized by recurrent translocations and somatic mutations. The prognosis of DLBCL has been associated with clinical features, cell-of-origin (COO) and genetic aberrations. The aim of this study was to determine whether somatic mutations are associated with overall survival (OS) in patients with DLBCL who have been treated with R-CHOP, and whether these mutations can be incorporated into a model to better predict survival.
Methods:
We identified 340 patients between 2000-2016 from two institutions with a diagnosis of de novo DLBCL treated with R-CHOP. A custom targeted panel with 334 genes which included frequently mutated genes in B-cell lymphoma was used and the captured DNA was sequenced on an Illumina HiSeq 2500. Cases were evaluated by immunohistochemistry (IHC: Hans algorithm; MYC and BCL2 expression), nCounter Nanostring (Lymph2Cx), and FISH analysis for BCL2, BCL6, and MYC rearrangement. OS was estimated using the Kaplan-Meier method. Multivariant modeling of OS was performed incorporating clinical features, IHC, COO by Nanostring, FISH, and mutation status of the most frequently mutated genes (≥5%). LASSO regression was performed to select for significant variables and determine coefficients for these variables and risk scores were calculated based on various fitted models. Concordance C-index was used to assess the discriminatory ability of different models, and three risk groups were determined by stratifying the risk scores in the final model.
Results:
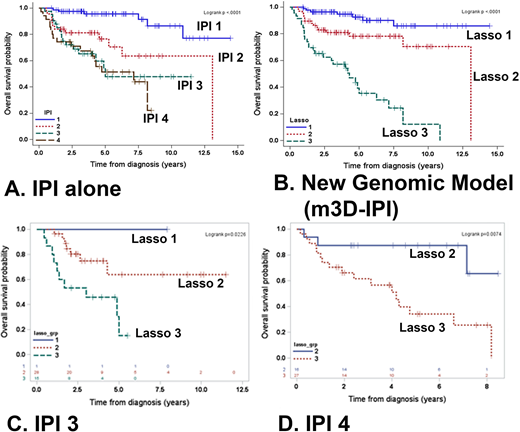
199 patients (median age 60 years, M:F ratio 1.4:1) had complete clinical and sequencing data. The germinal center B-cell phenotype (GCB) was more common (69%) than the activated B-cell phenotype (ABC; 26%), and 5% were unclassified by COO. The most frequently mutated genes were KMT2D (31%), CREBBP (21%), and TP53 (20%). Double/triple-hit (DH) lymphomas comprised of 11% of the cases, and 13% were double-protein expressors (MYC and BCL2) only. Significant variables selected by LASSO included factors in the IPI, FISH analysis, and 3 mutated genes (KMT2D, PIM1, and MEF2B). A formula was developed using the individual factors (elevated LDH, age ≥60 years, presence of extranodal sites, stage ≥ 3, male, DH status, and mutations in KMT2D, PIM1, and/or MEF2B. A three risk group model (m3D-IPI) was constructed based on these significant variables and its coefficients were superior in discriminating OS compared to the IPI alone (C-index: 0.830 vs. 0.775; Figures A and B). Within IPI group 3 (Figure C), Lasso 3 (high risk) identified patients that had a poor prognosis (p=0.022). In IPI group 4 (Figure D), Lasso 2 (intermediate risk) identified patients that had a better prognosis (p=0.0074). A simplified risk model using the same variables was also developed by assigning one point for each variable present, and these findings were validated in an independent cohort of DLBCL (Reddy et al, Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 2017;171(2):481-94).
Conclusion:
In this study, we incorporated mutation analysis of select genes with clinical risk factors and developed an improved risk model for patients with DLBCL treated with first-line therapy.
Herrera:Pharmacyclics: Consultancy, Research Funding; Gilead Sciences: Research Funding; Merck, Inc.: Consultancy, Research Funding; AstraZeneca: Research Funding; KiTE Pharma: Consultancy, Research Funding; Seattle Genetics: Research Funding; Bristol-Myers Squibb: Consultancy, Research Funding; Genentech: Consultancy, Research Funding; Immune Design: Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal